Targeted Sequencing (TAR) Reports
Djerba INI configuration
First, set up the working directory as outlined in Set up a Djerba working directory.
Information is usually obtained either from one of two Data Sources: the Requisition (Req) system or Dimsum.
The following information must be populated into the .ini file:
Parameter |
Source |
Description |
Example |
|---|---|---|---|
|
Dimsum, first link |
LIMS ID comprising the study name and patient number |
REVOLVE_0001 |
|
Dimsum, first link |
Name of the project in provenance |
REVTAR |
|
Req system, “Name of study (acronym)” under “Submission” tab |
Requisition system |
Re-VOLVE |
|
Req system, “OncoTree code” under “Submission” tab |
OncoTree code |
HGSOC |
|
(When not known, same as project) |
REVOLVE |
|
|
Req system. “Patient study ID” under “Submission” tab. |
Patient study ID in requisition system. Refer to the “External Names” in MISO to find the “Patient Study ID” within the requisition system, eg. REVOLVE_001 -> REV-TAR-329. |
REV-01-001 |
|
Dimsum, Test, Tumour TS |
ID of tumour sample |
REV-01-001_Pl |
|
Dimsum, Test, Normal TS |
ID of blood sample |
REV-01-001_BC |
|
Req system, “Primary cancer diagnosis” under “Submission” tab |
Name of primary cancer |
High grade serous ovarian carcinoma |
|
Req system |
Site of biopsy/surgery (usually cfDNA) |
cfDNA |
|
Req system |
Sample type (usually cfDNA) |
cfDNA |
|
Req system |
A known variant from previous genetic testing |
TP53 p.(D158*) |
|
Req system, ‘Submission approved” date under “Case History” tab |
Date of first requisition approval by Tissue Portal staff in yyyy-mm-dd format |
2023-10-31 |
|
Req system, Top of the requisition after “ID” |
Name of the requisition |
REVWGTS-P-861 |
|
Req system |
The assay used (targeted sequencing assay, value is “TAR”) |
TAR |
|
Dimsum - Full Depth Sequencings |
Default value is None |
REVOLVE_0001_01_LB01-02 |
|---|---|---|---|
|
Dimsum - Full Depth Sequencings |
Default value is None |
REVOLVE_0001_04_LB01-02 |
|
Dimsum - Full Depth Sequencings |
Default value is None |
REVOLVE_0001_04_LB01-01 |
|
Dimsum - Full Depth Sequencings |
REVOLVE_0001_01_LB01-02 |
|
Upon review of ichorCNA plot |
Default value is False |
False/True |
|---|---|---|---|
|
Upon review of the reported SNVs |
Default value is False |
False/True |
Example of a completed Djerba INI file
Spaces are acceptable in the parameter value and on either side of the = sign:
[core]
[tar_input_params_helper]
donor=REVOLVE_0001
project=REVTAR
study=Re-VOLVE
oncotree_code=HGSOC
cbio_id=REVOLVE
patient_study_id=REV-01-001
tumour_id=REV-01-001_Pl
normal_id=REV-01-001_BC
primary_cancer=High grade serous ovarian carcinoma
site_of_biopsy=cfDNA
sample_type = cfDNA
known_variants=<em>TP53</em> (p.D148*)
requisition_approved=2023-05-09
requisition_id = REVWGTS-P-861
assay=TAR
[provenance_helper]
sample_name_normal = REVOLVE_0001_01_LB01-02
sample_name_tumour = REVOLVE_0001_04_LB01-02
sample_name_aux = REVOLVE_0001_04_LB01-01
[report_title]
[patient_info]
[case_overview]
[gene_information_merger]
[treatment_options_merger]
[summary]
[tar.sample]
[tar.snv_indel]
[tar.swgs]
[tar.status]
copy_number_ctdna_detected = False
small_mutation_ctdna_detected = False
[supplement.body]
Interim Report Generation
Login and setup the analysis environment on a Univa compute node, as described in step 1.
Run djerba.py in report mode to generate an HTML report. (See below for examples.)
Output filename is of the form
${TUMOUR_ID}-v{VERSION_NUMBER}.htmlin the report directory, where $TUMOUR_ID is the tumour ID from Dimsum.Run the script using the INI file completed in Djerba INI configuration and the ‘report’ subdirectory created in Set up a Djerba working directory for intermediate output. Example:
$ djerba.py report -i config.ini -o report/
Proceed to review and interpretation of the interim HTML output.
Interpreting the TAR Report
Most results are reviewed within the interim report. Results reviewed by other means are explicitly mentioned in the text.
Review and confirm accuracy of non-PHI fields on interim report relative to current requisition in requisition portal in the case overview section.
Information regarding the tumour will be listed in the “Sample Information” section:
Review whizbam links for variants:
All variant calls must be viewed to gauge whether they are confident and thus reportable or an artifact and thus must be removed.
In general, if there are non-variant supporting reads in the normal, the variant is more likely to be an artifact.
Examples: Interpreting variants in Whizbam
Examine the copy number solution in
report/$(sample_name_aux)_genomeWide.pdf.If the tumour fraction is less than 10%, confirm that the plot is centered at 0 (i.e. that the majority of chromosomes have a log2ratio of 0, depicted by blue-shaded segments). If the tumour fraction is greater than 10%, confirm that the plot is centered at 0 and assess whether the elevated tumour fraction is driven by potentially artifactual chromosomal regions. Regions commonly observed as recurrent artifacts in healthy controls (i.e. likely false positives) include: 1p, 10q, 17, 19, and 22.
Examples: Interpreting ichorCNA CNV plots
If the elevated tumour fraction is likely driven by artifactual chromosomal regions, set the estimated tumour fraction to <10%. Copy number variants must be removed.
Subsequent time points to a positive:
For samples from a subsequent time point with tumour fraction <10%, compare the copy number profile to the preceding positive (>10%) time point. If a similar signal is present, retain the tumour fraction as <10% but report the sample as ctDNA detected (copy number analysis).
After reviewing both the copy number variants and the small mutations, the parameters in
[tar.status]in theconfig.inimay need to be adjusted. Bothcopy_number_ctdna_detectedandsmall_mutation_ctdna_detectedautomatically default to False.
After reviewing the SNVs and purity/CNVs, adjust the parameters as follows:
copy_number_ctdna_detected = Trueif the purity is ≥ 10%small_mutation_ctdna_detected = Trueif there are high confidence SNVs present
Once done, re-generate the report to ensure changes to
[tar.status]are rendered correctly:$ djerba.py report -i config.ini -o report/
For example, for a report with
copy_number_ctdna_detected = Trueandsmall_mutation_ctdna_detected = False, the output will be: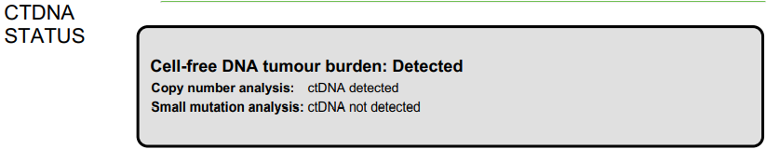
Once the variants to remove have been identified, remove them from
djerba_report.json. Working with JSON and DjerbaNote
For all follow-up cases, ensure that the status is consistent with the previous submission. If the case is positive — either due to a tumor fraction >10% or the presence of a reported SNV — be sure to double-check the original ichorCNA plot and confirm the variants reported in the initial submission.
It is not uncommon for the follow-up report to show new variants or higher tumor fraction; this can occur if the original sample was below our limit of detection. In such cases, review the old data in IGV to see if any supporting reads were present, and examine the ichorCNA plot for amplifications that may align with the current findings.
If prior knowledge of previous sequencing results or biomarkers is known, review the relevant sections of the report to confirm and note abnormalities.
If any discrepancy is noted, the sample should be marked as failed in Dimsum according to the QM-036 Quality Control Approval Procedure SOP and a Failed TAR Reports should be generated.
Table 13 Possible abnormalities Abnormality
Potential Cause
Action
Lack of expected alteration, or presence of a mutation in a cancer type where the mutation is expected or not expected
Lack of coverage for the expected mutation
Sample swap
Mutation is filtered
Verify coverage for the region by inspecting the bam file in Whizbam
Check for sample swaps
Confirm mutation was not removed by pipeline by reviewing the MuTect2 VCF file
Prior sequencing results are not confirmed
Low coverage for the expected mutation
Sample swap
Mutation is filtered
Verify coverage for the region by inspecting the bam file in Whizbam
Check for sample swaps
Confirm mutation was not removed by pipeline by reviewing the MuTect2 VCF file
Review the Small Mutations (SNVs/INDELs) section of the report. SNVs and INDELs are reported according to the following filtering criteria:
Table 14 SNV/InDels filter criteria Filter
Threshold
Variant Allele Frequency (VAF)
≥ 1%
Supporting Alternate Reads
≥ 3 reads
OncoKB
All level 1-4, R variants which pass the above criteria
All “Oncogenic”, “Likely Oncogenic” and “Predicted Oncogenic” alterations which pass the above criteria
Review all actionable and/or oncogenic mutations using Whizbam links for alignment artifacts. Whizbam links can be navigated to by clicking the link in the rightmost column in the
data_mutations_extended_oncogenic.txtfile in the patient’sreportdirectory. Alterations which are deemed artifacts are to be removed from the JSON file and recorded on the relevant JIRA ticket.Dinucleotide substitutions which are represented as two individual mutations are to be merged. Merged variants should be recorded in a new file named
data_mutations_merged.txt. Copy both original individual annotations to this file, along with a third record of the final merged variant. To perform this merge, please follow this step-by-step procedure in the Merging and Annotating Mutations Representing the Same Event🔒 document on CGI:How-to wiki page.
Generate an interpretation statement based on the findings from above and record it in a TXT file named
results_summary.txtFor samples flagged as follow-up, include an additional statement to comment on the shared and/or exclusive variants relative to prior sequencing results.
For an example summary, please refer to our wiki page on writing a genome interpretive statement🔒.
Use the following template as an example:
Table 15 Writing a TAR interpretation statement Analysis Subsection
Example statement
Comparison to prior sequencing results (for follow-up samples only)
Comment on the number of shared and exclusive mutations relative to prior sequencing results. When newly reported variants are discovered, include OncoKB recommendations for any new indications: “Relative to prior sequencing of [current sample X], [prior sample Y] shares 3 common variants and one variant is exclusive to, and has 1 additional oncogenic variant in gene A
SNV/Indel
“Mutations analysis uncovered loss of function mutations in xxx genes that suggest xxx.”
Copy Number
“Copy Number analysis uncovered an amplification in xxx genes that suggest xxx.”
OncoKB treatment recommendations
Statements are taken from oncoKB: “Alteration xxx is a Level 1 mutation which the following treatment recommendations according to oncoKB”
Updating QCs
The only QC for CGI to complete is to review the ichorCNA plot, which was done above.
Sign off on the “Informatics Review” in Dimsum according to the QM-036. Quality Control Approval Procedure.
Draft Report
Regenerate the PDF report with the interpretation changes and summary text:
Update the genomic summary text in the report JSON document as follows (note that input and output for the
update_summary.pyscript may be the same file):$ djerba.py update -s report/results_summary.txt -j report/report.json -o report/ -p
Continue to Review the Draft Report ➡️
Example Djerba TAR session
The following is an example sequence of commands used to generate a clinical report with Djerba. It is intended as a guide to CGI staff for report generation. The commands are for illustration only, not a fixed script to be followed. The start of each command is prefixed with $, and comments are prefixed with #:
$ ssh ugehn.hpc
$ sudo -u svc.cgiprod -i
$ qrsh -P gsi -l h_vmem=16G
$ module load djerba
$ cd WORK_DIR
# make a folder with the donor name, ex. REVOLVE_0001
$ mkdir REVOLVE_0001
$ cd REVOLVE_0001
# make a folder with the report directory, i.e. report/
$ mkdir report
# create a config.ini file
$ djerba.py setup --assay ASSAY --ini {WORK_DIR}/config.ini --compact –p ../../../CHARM2PLAS_project.ini
$ vim {WORK_DIR}/config.ini
# run djerba.py to generate a report
$ djerba.py report -i config.ini -o report/
# review the HTML
# review whizbam links in data_mutations_extended_oncogenic.txt
# remove any false calls in djerba_report.json (use json.tool to make it easier)
$ cat djerba_report.json | python3 -m json.tool > report/djerba_report_machine.pretty.json
$ vim djerba_report_machine.pretty.json
# edit results_summary.txt to write the genomic summary
$ vim report/results_summary.txt
# update the ctDNA plugin status from "Not Detected” to “Detected” if needed
# update the genomic summary
$ djerba.py update -s report/results_summary.txt -j report/report.json -o report/ -p
Change Log
June CR-119 update (#14) by Morgan Taschuk at 2026-07-21 19:22:33
* Remove references to mini-Djerba * Improve changelogs by pulling in the actual changes for each page with sphinx-git
Update tar-report.rst (#12) by Aqsa Alam at 2026-06-11 18:45:20
Update tar-report.rst with formatting changes by Aqsa Alam at 2026-06-05 17:55:29
Update tar-report.rst by Aqsa Alam at 2026-06-05 17:52:59
Add changelogs to every page; add a note about identical amended reports for CAPA-221 by Morgan Taschuk at 2025-09-15 20:10:46
Reorganized so each SOP has its own section by Morgan Taschuk at 2025-08-18 19:08:34